Chemical
- class thermosteam.Chemical(ID, cache=None, *, search_ID=None, eos=None, phase_ref=None, CAS=None, default=False, phase=None, search_db=True, V=None, Cn=None, mu=None, Cp=None, rho=None, sigma=None, kappa=None, epsilon=None, Psat=None, Hvap=None, **data)[source]
Creates a Chemical object which contains constant chemical properties, as well as thermodynamic and transport properties as a function of temperature and pressure.
- Parameters
ID (str) –
- One of the following [-]:
Name, in IUPAC form or common form or a synonym registered in PubChem
InChI name, prefixed by ‘InChI=1S/’ or ‘InChI=1/’
InChI key, prefixed by ‘InChIKey=’
PubChem CID, prefixed by ‘PubChem=’
SMILES (prefix with ‘SMILES=’ to ensure smiles parsing)
CAS number
cache (optional) – Whether or not to use cached chemicals and cache new chemicals.
search_ID (str, optional) – ID to search through database. Pass this key-word argument when you’d like to give a custom name to the chemical, but cannot find the chemical with that name.
eos (GCEOS subclass, optional) – Equation of state class for solving thermodynamic properties. Defaults to Peng-Robinson.
phase_ref ({'s', 'l', 'g'}, optional) – Reference phase of chemical.
CAS (str, optional) – CAS number of chemical.
phase ({'s', 'l' or 'g'}, optional) – Phase to set state of chemical.
search_db=True (bool, optional) – Whether to search the data base for chemical.
Cn (float or function(T), optional) – Molar heat capacity model [J/mol] as a function of temperature [K].
sigma (float or function(T), optional) – Surface tension model [N/m] as a function of temperature [K].
epsilon (float or function(T), optional) – Relative permitivity model [-] as a function of temperature [K].
Psat (float or function(T), optional) – Vapor pressure model [N/m] as a function of temperature [K].
Hvap (float or function(T), optional) – Heat of vaporization model [J/mol] as a function of temperature [K].
V (float or function(T, P), optional) – Molar volume model [mol/m3] as a function of temperature [K] and pressure [Pa].
mu (float or function(T, P), optional) – Dynamic viscosity model [Pa*s] as a function of temperature [K] and pressure [Pa].
kappa (float or function(T, P), optional) – Thermal conductivity model [W/m/K] as a function of temperature [K] and pressure [Pa].
Cp (float, optional) – Constant heat capacity model [J/g].
rho (float, optional) – Constant density model [kg/m3].
default=False (bool, optional) – Whether to default any missing chemical properties such as molar volume, heat capacity, surface tension, thermal conductivity, and molecular weight to that of water (on a weight basis).
**data (float or str) – User data (e.g. Tb, formula, etc.).
Examples
Chemical objects contain pure component properties:
>>> import thermosteam as tmo >>> # Initialize with an identifier >>> # (e.g. by name, CAS, InChI...) >>> Water = tmo.Chemical('Water') >>> Water.show() Chemical: Water (phase_ref='l') [Names] CAS: 7732-18-5 InChI: H2O/h1H2 InChI_key: XLYOFNOQVPJJNP-U... common_name: water iupac_name: ('oxidane',) pubchemid: 962 smiles: O formula: H2O [Groups] Dortmund: <1H2O> UNIFAC: <1H2O> PSRK: <1H2O> [Data] MW: 18.015 g/mol Tm: 273.15 K Tb: 373.12 K Tt: 273.15 K Tc: 647.14 K Pt: 610.88 Pa Pc: 2.2048e+07 Pa Vc: 5.6e-05 m^3/mol Hf: -2.8582e+05 J/mol S0: 70 J/K/mol LHV: 44011 J/mol HHV: 0 J/mol Hfus: 6010 J/mol Sfus: None omega: 0.344 dipole: 1.85 Debye similarity_variable: 0.16653 iscyclic_aliphatic: 0 combustion: {'H2O': 1.0}
All fields shown are accessible:
>>> Water.CAS '7732-18-5'
Functional group identifiers (e.g. Dortmund, UNIFAC, PSRK) allow for the estimation of activity coefficients through group contribution methods. In other words, these attributes define the functional groups for thermodynamic equilibrium calculations:
>>> Water.Dortmund <DortmundGroupCounts: 1H2O>
Temperature (in Kelvin) and pressure (in Pascal) dependent properties can be computed:
>>> # Vapor pressure (Pa) >>> Water.Psat(T=373.15) 101284.55... >>> # Surface tension (N/m) >>> Water.sigma(T=298.15) 0.07197220523... >>> # Molar volume (m^3/mol) >>> Water.V(phase='l', T=298.15, P=101325) 1.80692...e-05
Note that the reference state of all chemicals is 25 degC and 1 atm:
>>> (Water.T_ref, Water.P_ref) (298.15, 101325.0) >>> # Enthalpy at reference conditions (J/mol; without excess energies) >>> Water.H(T=298.15, phase='l') 0.0
Constant pure component properties are also available:
>>> # Molecular weight (g/mol) >>> Water.MW 18.01528 >>> # Boiling point (K) >>> Water.Tb 373.124
Temperature dependent properties are managed by model handles:
>>> Water.Psat.show() TDependentModelHandle(T, P=None) -> Psat [Pa] [0] Wagner original [1] Antoine [2] EQ101 [3] Wagner [4] boiling critical relation [5] Lee Kesler [6] Ambrose Walton [7] Sanjari [8] Edalat
Phase dependent properties have attributes with model handles for each phase:
>>> Water.V <PhaseTPHandle(phase, T, P) -> V [m^3/mol]> >>> (Water.V.l, Water.V.g) (<TPDependentModelHandle(T, P) -> V.l [m^3/mol]>, <TPDependentModelHandle(T, P) -> V.g [m^3/mol]>)
A model handle contains a series of models applicable to a certain domain:
>>> Water.Psat[0].show() TDependentModel(T, P=None) -> Psat [Pa] name: Wagner original Tmin: 275 K Tmax: 647.35 K
When called, the model handle searches through each model until it finds one with an applicable domain. If none are applicable, a value error is raised:
>>> Water.Psat(373.15) 101284.55179999319 >>> # Water.Psat(1000.0) -> >>> # ValueError: Water has no valid saturated vapor pressure model at T=1000.00 K
Model handles as well as the models themselves have tabulation and plotting methods to help visualize how properties depend on temperature and pressure.
>>> # import matplotlib.pyplot as plt >>> # Water.Psat.plot_vs_T([Water.Tm, Water.Tb], 'degC', 'atm', label="Water") >>> # plt.show()
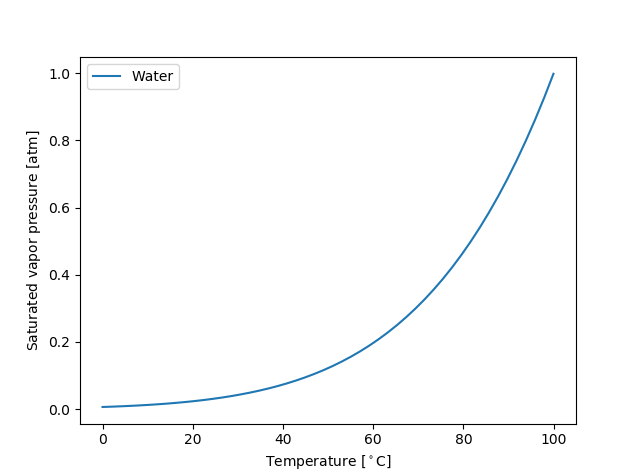
>>> # Plot all models
>>> # Water.Psat.plot_models_vs_T([Water.Tm, Water.Tb], 'degC', 'atm') >>> # plt.show()
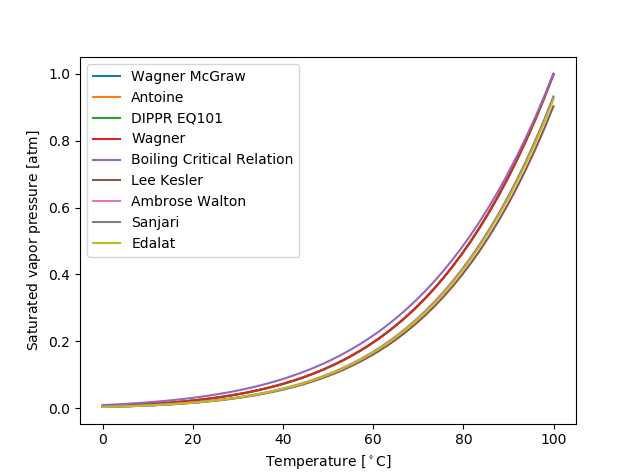
Each model may contain either a function or a functor (a function with stored data) to compute the property:
>>> functor = Water.Psat[0].evaluate >>> functor.show() Functor: Wagner_original(T, P=None) -> Psat [Pa] Tc: 647.35 K Pc: 2.2122e+07 Pa a: -7.7645 b: 1.4584 c: -2.7758 d: -1.233
Note
All chemical property functors are available in the thermosteam.properties subpackage. You can also use help(<functor>) for further information on the math and equations used in the functor.
A new model can be added easily to a model handle through the add_model method, for example:
>>> # Set top_priority=True to place model in postion [0] >>> @Water.Psat.add_model(Tmin=273.20, Tmax=473.20, top_priority=True) ... def User_antoine_model(T): ... return 10.0**(10.116 - 1687.537 / (T - 42.98)) >>> Water.Psat.show() TDependentModelHandle(T, P=None) -> Psat [Pa] [0] User antoine model [1] Wagner original [2] Antoine [3] EQ101 [4] Wagner [5] boiling critical relation [6] Lee Kesler [7] Ambrose Walton [8] Sanjari [9] Edalat
The add_model method is a high level interface that even lets you create a constant model:
>>> value = Water.V.l.add_model(1.687e-05, name='User constant') ... # Model is appended at the end by default >>> added_model = Water.V.l[-1] >>> added_model.show() ConstantThermoModel(T=None, P=None) -> V.l [m^3/mol] name: User constant value: 1.687e-05 Tmin: 0 K Tmax: inf K Pmin: 0 Pa Pmax: inf Pa
Note
Because no bounds were given, the model assumes it is valid across all temperatures and pressures.
Manage the model order with the set_model_priority and move_up_model_priority methods:
>>> # Note: In this case, we pass the model name, but its >>> # also possible to pass the current index, or the model itself. >>> Water.Psat.move_up_model_priority('Wagner original') >>> Water.Psat.show() TDependentModelHandle(T, P=None) -> Psat [Pa] [0] Wagner original [1] Antoine [2] EQ101 [3] Wagner [4] boiling critical relation [5] Lee Kesler [6] Ambrose Walton [7] Sanjari [8] Edalat [9] User antoine model
>>> Water.Psat.set_model_priority('Antoine') >>> Water.Psat.show() TDependentModelHandle(T, P=None) -> Psat [Pa] [0] Antoine [1] Wagner original [2] EQ101 [3] Wagner [4] boiling critical relation [5] Lee Kesler [6] Ambrose Walton [7] Sanjari [8] Edalat [9] User antoine model
The default priority is 0 (or top priority), but you can choose any priority:
>>> Water.Psat.set_model_priority('Antoine', 1) >>> Water.Psat.show() TDependentModelHandle(T, P=None) -> Psat [Pa] [0] Wagner original [1] Antoine [2] EQ101 [3] Wagner [4] boiling critical relation [5] Lee Kesler [6] Ambrose Walton [7] Sanjari [8] Edalat [9] User antoine model
Note
All models are stored as a
dequein the models attribute (e.g. Water.Psat.models).- mu(phase, T, P)
Dynamic viscosity [Pa*s].
- kappa(phase, T, P)
Thermal conductivity [W/m/K].
- V(phase, T, P)
Molar volume [m^3/mol].
- Cn(phase, T)
Molar heat capacity [J/mol/K].
- Psat(T)
Vapor pressure [Pa].
- Hvap(T)
Heat of vaporization [J/mol]
- sigma(T)
Surface tension [N/m].
- epsilon(T)
Relative permitivity [-]
- S(phase, T, P)
Entropy [J/mol].
- H(phase, T)
Enthalpy [J/mol].
- S_excess(T, P)
Excess entropy [J/mol].
- H_excess(T, P)
Excess enthalpy [J/mol].
- T_ref = 298.15
[float] Reference temperature in Kelvin
- P_ref = 101325.0
[float] Reference pressure in Pascal
- H_ref = 0.0
[float] Reference enthalpy in J/mol
- chemical_cache = {}
dict[str, Chemical] Cached chemicals
- cache = False
[bool] Wheather or not to search cache by default
- classmethod new(ID, CAS, eos=<class 'thermosteam.eos.PR'>, phase_ref=None, phase=None, **data)[source]
Create a new chemical from data without searching through the data base, and load all possible models from given data.
- classmethod blank(ID, CAS=None, phase_ref=None, phase=None, formula=None, **data)[source]
Return a new Chemical object without any thermodynamic models or data (unless provided).
- Parameters
ID (str) – Chemical identifier.
CAS (str, optional) – CAS number. If none provided, it defaults to the ID.
phase_ref (str, optional) – Phase at the reference state (T=298.15, P=101325).
phase (str, optional) – Phase to set state as a single phase chemical.
**data – Any data to fill chemical with.
Examples
>>> from thermosteam import Chemical >>> Substance = Chemical.blank('Substance') >>> Substance.show() Chemical: Substance (phase_ref=None) [Names] CAS: Substance InChI: None InChI_key: None common_name: None iupac_name: None pubchemid: None smiles: None formula: None [Groups] Dortmund: <Empty> UNIFAC: <Empty> PSRK: <Empty> [Data] MW: None Tm: None Tb: None Tt: None Tc: None Pt: None Pc: None Vc: None Hf: None S0: None LHV: None HHV: None Hfus: None Sfus: None omega: None dipole: None similarity_variable: None iscyclic_aliphatic: None combustion: None
- copy(ID, CAS=None, **data)[source]
Return a copy of the chemical with a new ID.
Examples
>>> import thermosteam as tmo >>> Glucose = tmo.Chemical('Glucose') >>> Mannose = Glucose.copy('Mannose') >>> Mannose.show() Chemical: Mannose (phase_ref='l') [Names] CAS: Mannose InChI: C6H12O6/c7-1-3(9)5(1... InChI_key: GZCGUPFRVQAUEE-S... common_name: D-Glucose iupac_name: ('(2R,3S,4R,5R)... pubchemid: 1.0753e+05 smiles: O=C[C@H](O)[C@@H](O... formula: C6H12O6 [Groups] Dortmund: {2: 1, 3: 4, 14: ... UNIFAC: {2: 1, 3: 4, 14: 5,... PSRK: {2: 1, 3: 4, 14: 5, 2... [Data] MW: 180.16 g/mol Tm: None Tb: 616.29 K Tt: None Tc: 755 K Pt: None Pc: 4.82e+06 Pa Vc: 0.000414 m^3/mol Hf: -1.2711e+06 J/mol S0: 0 J/K/mol LHV: -2.5406e+06 J/mol HHV: -2.8047e+06 J/mol Hfus: 0 J/mol Sfus: None omega: 2.387 dipole: None similarity_variable: 0.13322 iscyclic_aliphatic: 0 combustion: {'CO2': 6, 'O2'...
- property phase_ref
{‘s’, ‘l’, ‘g’} Phase at 298 K and 101325 Pa.
- property ID
[str] Identification of chemical.
- property CAS
[str] CAS number of chemical.
- property InChI
[str] IUPAC International Chemical Identifier.
- property InChI_key
[str] IUPAC International Chemical Identifier shorthand.
- property common_name
[str] Common name identifier.
- property iupac_name
[str] Standard name as defined by IUPAC.
- property pubchemid
[str] Chemical identifier as defined by PubMed.
- property smiles
[str] Chemical SMILES formula.
- property formula
[str] Chemical atomic formula.
- property Dortmund
[DortmundGroupCounts] Dictionary-like object with functional group numerical identifiers as keys and the number of groups as values.
- property UNIFAC
[UNIFACGroupCounts] Dictionary-like object with functional group numerical identifiers as keys and the number of groups as values.
- property PSRK
[PSRKGroupCounts] Dictionary-like object with functional group numerical identifiers as keys and the number of groups as values.
- property eos
[object] Instance for solving equations of state.
- property eos_1atm
[object] Instance for solving equations of state at 1 atm.
- property mu
Dynamic viscosity [Pa*s].
- property kappa
Thermal conductivity [W/m/K].
- property V
Molar volume [m^3/mol].
- property Cn
Molar heat capacity [J/mol/K].
- property Psat
Vapor pressure [Pa].
- property Hvap
Heat of vaporization [J/mol].
- property sigma
Surface tension [N/m].
- property epsilon
Relative permitivity [-].
- property S
Entropy [J/mol].
- property H
Enthalpy [J/mol].
- property S_excess
Excess entropy [J/mol].
- property H_excess
Excess enthalpy [J/mol].
- property MW
Molecular weight [g/mol].
- property Tm
Normal melting temperature [K].
- property Tb
Normal boiling temperature [K].
- property Pt
Triple point pressure [Pa].
- property Tt
Triple point temperature [K].
- property Tc
Critical point temperature [K].
- property Pc
Critical point pressure [Pa].
- property Vc
Critical point molar volume [m^3/mol].
- property Hfus
Heat of fusion [J/mol].
- property Sfus
Entropy of fusion [J/mol].
- property omega
Accentric factor [-].
- property dipole
Dipole moment [Debye].
- property similarity_variable
Similarity variable [-].
- property iscyclic_aliphatic
Whether the chemical is cyclic-aliphatic.
- property Hf
Heat of formation at reference phase and temperature [J/mol].
- property LHV
Lower heating value [J/mol].
- property HHV
Higher heating value [J/mol].
- property combustion
dict[str, int] Combustion reaction.
- property Stiel_Polar
[float] Stiel Polar factor for computing surface tension.
- property Zc
[float] Compressibility factor.
- property has_hydroxyl
[bool] Whether or not chemical contains a hydroxyl functional group, as determined by the Dortmund/UNIFAC/PSRK functional groups.
- property atoms
int] Atom-count pairs based on formula.
- Type
dict[str
- get_combustion_reaction(chemicals=None)[source]
Return a Reaction object defining the combustion of this chemical. If no combustion stoichiometry is available, return None.
- get_phase(T=298.15, P=101325.0)[source]
Return phase of chemical at given thermal condition.
Examples
>>> from thermosteam import Chemical >>> Water = Chemical('Water', cache=True) >>> Water.get_phase(T=400, P=101325) 'g'
- Tsat(P, Tguess=None, Tmin=None, Tmax=None)[source]
Return the saturated temperature (in Kelvin) given the pressure (in Pascal).
- reset(CAS, eos=<class 'thermosteam.eos.PR'>, phase_ref=None, smiles=None, InChI=None, InChI_key=None, pubchemid=None, iupac_name=None, common_name=None, formula=None, MW=None, Tm=None, Tb=None, Tc=None, Pc=None, Vc=None, omega=None, Tt=None, Pt=None, Hf=None, S0=None, LHV=None, combustion=None, HHV=None, Hfus=None, dipole=None, similarity_variable=None, iscyclic_aliphatic=None, *, metadata=None, phase=None)[source]
Reset all chemical properties.
- Parameters
CAS (str) – CAS number of chemical to load.
eos (optional) – Equation of state. The default is Peng Robinson.
phase_ref (str, optional) – Phase reference. Defaults to the phase at 298.15 K and 101325 Pa.
- reset_combustion_data(method='Stoichiometry')[source]
Reset combustion data (LHV, HHV, and combution attributes) based on the molecular formula and the heat of formation.
- get_key_property_names()[source]
Return the attribute names of key properties required to model a process.
- default(properties=None)[source]
Default all properties with the chemical properties of water. If no properties given, all essential chemical properties that are missing are defaulted. properties which are still missing are returned as set.
- Parameters
properties (Iterable[str], optional) – Names of chemical properties to default.
- Returns
missing_properties – Names of chemical properties that are still missing.
- Return type
set[str]
Examples
>>> from thermosteam import Chemical >>> Substance = Chemical.blank('Substance') >>> missing_properties = Substance.default() >>> sorted(missing_properties) ['Dortmund', 'Hfus', 'Hvap', 'PSRK', 'Pc', 'Psat', 'Pt', 'Sfus', 'Tb', 'Tc', 'Tm', 'Tt', 'UNIFAC', 'V', 'Vc', 'dipole', 'iscyclic_aliphatic', 'omega', 'similarity_variable']
Note that missing properties does not include essential properties volume, heat capacity, and conductivity.
- get_missing_properties(properties=None)[source]
Return a list all missing thermodynamic properties.
Examples
>>> from thermosteam import Chemical >>> Substance = Chemical.blank('Substance', phase_ref='l') >>> sorted(Substance.get_missing_properties()) ['Cn', 'Dortmund', 'H', 'HHV', 'H_excess', 'Hf', 'Hfus', 'Hvap', 'LHV', 'MW', 'PSRK', 'Pc', 'Psat', 'Pt', 'S', 'S_excess', 'Sfus', 'Tb', 'Tc', 'Tm', 'Tt', 'UNIFAC', 'V', 'Vc', 'combustion', 'dipole', 'epsilon', 'iscyclic_aliphatic', 'kappa', 'mu', 'omega', 'sigma', 'similarity_variable']
- property locked_state
[str] Constant phase of chemical.